

Abstract
Scaffolding proteins represent an evolutionary solution to controlling the specificity of information transfer in intracellular networks. They are highly concentrated in complexes located in specific subcellular locations. One of these complexes is the postsynaptic density of the excitatory synapses. There, scaffolding proteins regulate various processes related to synaptic plasticity, such as glutamate receptor trafficking and signalling, and dendritic structure and function. Most scaffolding proteins can be grouped into 4 main families: discs large (DLG), discs-large-associated protein (DLGAP), Shank and Homer. Owing to the importance of scaffolding proteins in postsynaptic density architecture, it is not surprising that variants in the genes that code for these proteins have been associated with neuropsychiatric diagnoses, including schizophrenia and autism-spectrum disorders. Such evidence, together with the clinical, neurobiological and genetic overlap described between schizophrenia and autism-spectrum disorders, suggest that alteration of scaffolding protein dynamics could be part of the pathophysiology of both. However, despite the potential importance of scaffolding proteins in these psychiatric conditions, no systematic review has integrated the genetic and molecular data from studies conducted in the last decade. This review has the following goals: to systematically analyze the literature in which common and/or rare genetic variants (single nucleotide polymorphisms, single nucleotide variants and copy number variants) in the scaffolding family genes are associated with the risk for either schizophrenia or autism-spectrum disorders; to explore the implications of the reported genetic variants for gene expression and/or protein function; and to discuss the relationship of these genetic variants to the shared genetic, clinical and cognitive traits of schizophrenia and autism-spectrum disorders.
Introduction
Schizophrenia and autism-spectrum disorders are neurodevelopmental psychiatric disorders that have a prevalence of approximately 1% and 2.5% worldwide, respectively,1,2 and have profound human and economic consequences.
Schizophrenia and autism-spectrum disorders were nosologically separated in the Diagnostic and Statistical Manual Mental Disorders, third edition (1980).3 However, evidence has been accumulating to suggest that they may partially overlap in their clinical, neurobiological, behavioural and cognitive features, and that they may have some common etiological roots.4 Regarding their clinical expression, some authors have proposed that the negative symptoms of schizophrenia can be construed more broadly as deficits in social communication and motivation, which are also found in people with autism-spectrum disorders.5 Similarly, the grossly disorganized or abnormal motor behaviour described in schizophrenia includes a number of signs and symptoms consistent with those of autism-spectrum disorders, such as repeated stereotyped movements, echolalia, unpredictable agitation and decreased interaction with or interest in one’s environment.5,6 The disorders also share some cognitive deficits7–9; in particular, deficits in social cognition have received much attention.10–15 As well, there are brain structural similarities between these disorders. For instance, lower grey matter volume in the limbic–striato–thalamic circuitry is common to schizophrenia and autism-spectrum disorders,16 and reduced volume and thickness of the insula have been found in patients with first-episode psychosis and in high-functioning patients with autism-spectrum disorders.17 Similar alterations to the white matter integrity of the left fronto-occipital fasciculus have recently been found in patients with schizophrenia and in patients with autism-spectrum disorders.18
In recent years, the field of molecular genetics has been uncovering evidence of an overlapping and complex polygenetic architecture for these disorders. Evidence suggests that studying pathways common to both may shed light on their pathophysiology and clinical heterogeneity.
Robust longitudinal and epidemiological studies have shown that 25% of people with childhood-onset schizophrenia have a history of a premorbid autism-spectrum disorder19; that the adult outcomes of children with atypical autism include psychotic disorders20; that autistic traits in infancy increase the risk for psychotic experiences later in life21; and that there is some co-occurrence of autism-spectrum disorders and psychotic disorders.22,23 This overlap is further supported by family studies, which have reported that the presence of one of these diagnoses in a first-degree relative increases the risk of the other.24–27 Similarly, schizophrenia is more common in parents of patients with autism than in parents of healthy controls.24
Twin studies have also recognized the important contribution of genetic factors to both schizophrenia and autism-spectrum disorders, with heritability estimates of h2 = 64%–80%28,29 and h2 = 64%–91%,30 respectively.
Over the last decade, molecular studies have contributed to our initial understanding of the complex genetic architecture of schizophrenia and autism-spectrum disorders, and later to identifying genes that are involved in both disorders. In this sense, it is currently accepted that an individual’s genetic risk of schizophrenia or an autism-spectrum disorder can be attributed to either many common variants with a frequency of > 1% (single nucleotide polymorphisms [SNPs]), each conferring a modest level of risk (odds ratio = 1.1–1.5); or rare mutations with a frequency of < 1% (single nucleotide variants [SNVs] and copy number variants [CNVs]) that are usually associated with a larger penetrance on the phenotype (odds ratio > 2).31,32
The most recent studies to examine genome-wide SNPs contributing to these disorders have estimated that genetic variation from SNPs accounts for 23% and 17% of the variance in risk of schizophrenia and autism-spectrum disorders, respectively.33 Based on the significant but small correlation between SNP heritability estimates in both disorders, the coheritability between them has been quantified at around 4%.33 In this regard, genome-wide association studies have identified several SNPs associated with schizophrenia and/or autism-spectrum disorders.34–37
Meanwhile, genome-wide and microarray-based comparative genomic hybridizations have found that the CNV burden is also increased in patients with schizophrenia or autism-spectrum disorders compared with healthy controls.38–41 For example, microduplications of 1q21.1 or 16p11.2 and deletions at 2p16.3, 15q11.2 or 22q11.21 have been reported in patients with schizophrenia and autism-spectrum disorders.42 De novo gene-disrupting SNVs have also been found to occur at higher rates in patients with autism-spectrum disorders than in controls.43–46 In schizophrenia, the initially reported increased rates of putatively functional mutations47,48 have not been replicated in 2 larger studies,49,50 but those later studies found that the enrichment of loss-of-function de novo mutations was relatively concentrated in genes that overlapped with those affected by de novo mutations in autism-spectrum disorders. In addition, an excess of de novo mutation was confirmed in an independent sample of patients with schizophrenia.51
With regard to the identification of specific genes involved in both schizophrenia and autism-spectrum disorders, findings from CNV and SNV studies have shown a notable consistency in some functionally enriched sets of genes. Genetic studies assessing common or rare variants show a certain convergence on reporting genes involved in glutamatergic synapse plasticity.49,52–55 A structure located in glutamatergic synapses that has been associated with both disorders is the postsynaptic density (PSD).50,56–61 For example, Bayés and colleagues found that mutations in 199 human PSD genes were involved in more than 200 diseases, half being nervous-system disorders.61 That study suggested that impairments in PSD proteins might underlie psychiatric disorders and their associated cognitive, behavioural and clinical phenotypes, but no systematic review based on this hypothesis has integrated the molecular data generated across studies in the last decade. Another example has been provided by Purcell and colleagues, who, after analyzing the exome sequences of 2536 patients with schizophrenia and 2543 controls, reported that SNVs were significantly more frequent in cases than controls, and that these SNVs were especially enriched in the activity-regulated cytoskeleton-associated (ARC) complex of PSD.50
PSD proteins and pathophysiological hypotheses
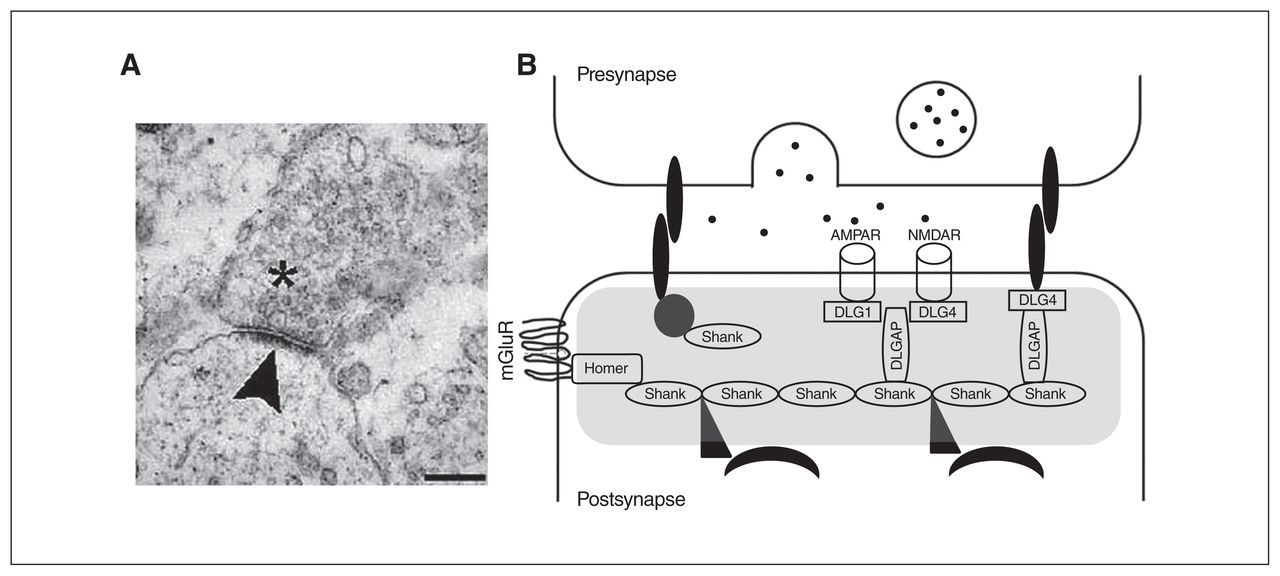
The PSD is a specialized matrix located at the excitatory postsynaptic terminals with a dish-shaped aspect, a surface area of 0.07 μm2 and a thickness of 30 to 40 nm on electron microscopy (Fig. 1A).62 The PSD can also be described as a highly organized and dynamic macromolecular complex consisting of several hundred proteins that process, integrate and converge the excitatory glutamatergic synaptic signals on the nucleus. As a point of convergence for the glutamatergic signalling pathways with other neurotransmitter systems, the composition and regulation of the PSD is essential for ensuring normal synaptic neurotransmission and plasticity.63,64

Image of the postsynaptic density (PSD) and scheme of the scaffolding proteins at the PSD that have been analyzed in the present review. (A) An electronic microscope image of a synapse; vesicles can be observed in the presynaptic neuron (asterisk). The electron-dense structure observed in the postsynaptic element is the PSD (arrowhead). Scale bar, 250 nm. Image retrieved under the Creative Commons Attribution License from Heupel et al. Neural Devel 2008, https://doi.org/10.1186/1749-8104-3-25. (B) A scheme of the PSD (grey shading). Multimerization of Shank1 to 3 proteins generate a network that links numerous proteins to the postsynaptic receptors. Homer proteins, including Homer1b/c, Homer2 and Homer3, also act as adaptors and interact with several PSD proteins, such as type I-mGluRs. The DLGAP1 to 4 proteins interact with DLG proteins, including the DLG1/SAP-97, DLG2/PSD-93, DLG3/SAP-102 and DLG4/PSD-95, to coregulate different ion channels, such as the NMDAR and AMPAR. AMPAR = α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; DLG = discs large; DLGAP = discs-large-associated protein; mGluR = metabotropic glutamate receptor; NMDAR = N-methyl-d-aspartate receptor; SAP = synapse-associated protein.
The PSD is enriched with different membrane components, such as glutamate receptors, tyrosine kinases, G protein–coupled receptors, ion channels or cell adhesion molecules, which are assembled by cytoplasmatic scaffolding proteins.65 Among the proteins that make up the PSD, studies have reported associations between genes coding for scaffolding proteins and both schizophrenia and autism-spectrum disorders, suggesting that variants in these genes might increase the risk of these disorders. For instance, recent studies have found that SNPs and CNVs in autism-spectrum disorders and schizophrenia are particularly concentrated in scaffolding genes and other PSD-related genes.66,67 Other studies have indicated changes in the expression of scaffolding genes in schizophrenia and autism-spectrum disorders compared with healthy controls.68,69 A recent study reported that gene-disrupting ultra-rare variants were more abundant in schizophrenia cases than in controls, and that these mutations were particularly enriched in scaffolding genes and other PSD genes.70
Scaffolding proteins can be defined as molecular circuit boards that can organize a wide variety of circuit relationships between signalling proteins. More specifically, the main function of scaffolding proteins is to bring together 2 or more proteins to facilitate their interaction and functions, linked to critical roles in cellular signalling. This is possible because scaffolding proteins are composed of several protein–protein interaction modules, most notably the PSD-95/discs large/zona occludens-1 (PDZ) and Src homology 3 (SH3) domains.71 Since scaffolding protein complexes are dynamic, they have the ability to change specific protein interactions to rapidly adapt to changing environmental requirements or diverse signalling cues.72 This versatility is related to their modularity, which allows for recombination of protein interaction domains to generate variability in signalling pathways. Such properties are seen as a simple evolutionary solution to controlling the specificity of information flow in intracellular networks, generating precise signalling behaviours.73
Owing to their dynamic configuration, postsynaptic scaffolding molecules not only establish the internal organization of the PSD, allowing neurons to respond efficiently to stimuli, but they also regulate processes related to synaptic plasticity, such as glutamate receptor trafficking and signalling, and dendritic structure and function,74,75 which critically determine the characteristics of excitatory synaptic transmission (Fig. 1B).
Disruption of scaffolding genes might alter the homeostasis of the PSD and contribute to the synaptic dysfunctions associated with schizophrenia and autism-spectrum disorders.76 However, despite the potential importance of scaffolding proteins in these psychiatric conditions, no systematic review has addressed the integration of genetic and molecular data generated across studies.
The nature and function of the different families of scaffolding proteins included in this review, and the characteristics of the genes encoding them, are shown in Figure 1 and briefly summarized below.
The discs large protein subfamily
The discs large (DLG) subfamily is a group of proteins in the membrane-associated guanylate kinase (MAGUK) family, and consists of DLG1, DLG2, DLG3 and DLG4. These proteins have 3 PDZ domains in their N-terminus, which allow them to interact with a variety of binding partners in the PSD, such as glutamatergic receptors, as well as other cytoplasmic scaffolding proteins. The DLG proteins control the transmission of extracellular signals to downstream signalling molecules of the PSD and regulate the localization of glutamatergic receptors N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) at neuronal synapses and dendrites.77 Moreover, they regulate the trafficking and clustering of ionic channels and the excitability of the presynaptic terminals, affecting the amount of neurotransmitter released.78 Since their temporal and spatial expression differ, it is believed that DLG members complement each other in performing these functions from embryonic to adult stages.79
The DLG1 gene (also known as SAP-97) maps on chromosome 3q29 and encodes the synapse-associated protein 97 (SAP-97 or DLG1), which is thought to play a role in synaptogenesis80 and glutamatergic receptor trafficking during development.77
The DLG2 gene (also known as PSD-93) is located on chromosome 11q14.1. Different studies have suggested that the protein it encodes (PSD-93, DLG2) plays a role in the regulation of synaptic plasticity. The DLG2 protein interacts with the tyrosine kinase Fyn, which is involved in the phosphorylation-based regulation of NMDA receptors that is required for the induction of NMDA-receptor-dependent long-term potentiation.81
The DLG3 gene (also known as SAP-102) is located on Xq13.1, and the protein it encodes (DLG3) is the first protein related to intellectual disability that has been directly linked to glutamate receptor signalling and trafficking.82 Later studies have replicated the association of this gene with intellectual disability,83,84 suggesting that DLG3 somehow modulates cognition. This is consistent with the observed embryonic expression of this protein and its role in the regulation of synaptic formation and plasticity during brain development.85
The DLG4 gene is located on chromosome 17p13.1, and the protein it encodes (PSD-95, DLG4) is involved in the maturation of synapse formation and the NMDA receptor signalling pathway. It participates in the clustering and trafficking of NMDA and AMPA receptors in the PSD.63,79 Moreover, DLG4 interacts with the dopamine receptor D1 (DRD1) and the NMDA receptor, and regulates positive feedback between them.86 The degradation of DLG4 is regulated by other proteins that have also been associated with autism-spectrum disorders.87
The discs-large-associated protein family
The discs-large-associated protein (DLGAP) family is made up of 4 proteins encoded by different homonymous genes: DLGAP1 (18p11), DLGAP2 (8p23), DLGAP3 (1p34), and DLGAP4 (20q11). All proteins have 5 repeats of 14 amino acids in the middle region, followed by a proline-rich sequence and a C-terminal PDZ-binding motif that mediate interactions with other PSD proteins.88
Although the differential roles of each family member are unknown, all DLGAP proteins play an important role in organizing the postsynaptic signalling complex in glutamatergic synapses,89 and are especially involved in the stabilization of synaptic junctions and regulation of neurotransmission.90 In addition, DLGAP proteins clearly have a central role in the regulation of synaptic ion channels, including both NMDA and AMPA receptors.91
DLGAP2 (also known as SAPAP2 or GKAP) is the most studied of the proteins in this family. It interacts directly with DLG4 and Shank proteins to form a complex that plays critical roles in synaptic morphogenesis and function.90,92
It has been proposed that SAPAP proteins provide a link between the PSD-95 family of proteins and the actincytoskeleton through interactions with the Shank/ProSAP proteins, which in turn bind the actin-binding protein cortactin.93–97 Additionally, Shank/ProSAP binds to Homer, which interacts with metabotropic glutamate receptors.98 Therefore, in the current scaffolding model, PSD-95/SAPAP/Shank interactions play an important role in organizing the large postsynaptic signalling complex at glutamatergic synapses.96,97,99,100
The Shank protein family
Another PSD scaffolding protein group is the SH3 and multiple ankyrin repeat domains (Shank) family, which consists of 3 proteins encoded by different genes that are differently expressed in the brain97,101: SHANK1 (19q13.33), SHANK2 (11q13.3) and SHANK3 (22q13.3). So far, it is unclear whether individual Shank family proteins fulfill unique physiologic functions, but the structural similarity between Shank forms has led to the observation that many interaction partners of Shank proteins in the synapse are recognized equally by all 3 family members.93
In this regard, Shank proteins crosslink Homer, DLGAP2 and DLG4 proteins in the PSD and participate in glutamatergic downstream signalling by assembling glutamate receptors with other scaffolding proteins, cytoskeleton factors and intracellular effectors.102 Multimerization of Shank1–3 proteins can generate a network in the PSD that links numerous proteins to the postsynaptic receptors. In addition, Shank proteins promote the formation, maturation and enlargement of dendritic spines.103
The Homer protein family
Homer proteins include 3 different members that are encoded by 3 different homonymous genes: HOMER1 (5q14), HOMER2 (15q25) and HOMER3 (19p13). Homer proteins can also be classified into constitutively expressed isoforms (i.e., Homer1b/c, Homer2 and Homer3), which are bimodal proteins with an N-terminal domain that mediates the interaction with other PSD proteins, and a C-terminal coiled-coil domain that enables self-assembly, as well as short splice variants (Homer1a and Ania-3) that lack the C-terminal domain and cannot self-assemble.104 The various protein forms are differentially expressed over time and place.105
Homer proteins act as multimodal adaptors by interacting with several PSD proteins, such as type I metabotropic glutamate receptors (mGLuR1–5), Shank proteins or synaptic signalling molecules such as inositol 1,4,5-triphosphate receptors (IP3Rs), and binding them to the cytoskeleton.102 Homer proteins are also involved in glutamatergic synapses by regulating glutamatergic receptor trafficking, the function of plasma membrane ion channels and intracellular messenger systems.106 For these reasons, Homer proteins are important for cell signalling, cell excitability, synaptic neurotransmission and neuronal plasticity.107,108
Objectives
The objectives of this review were as follows: to conduct a systematic review of the literature in which variations within the above-mentioned scaffolding genes are associated with either schizophrenia or autism-spectrum disorders, and to describe the degree of overlap between both diagnoses; to explore whether the reported genetic variants putatively associated with schizophrenia or autism-spectrum disorders are involved in changes of gene expression or protein functionality according to basic research data; and to consider the implications of the reported associations for the development of these disorders and their associated phenotypes.
Methods
We conducted a systematic search of the PubMed, PsycINFO and Web of Science databases. The search terms were “Schizophrenia or autism” and “postsynaptic density proteins or PSD or scaffold* proteins” without date restrictions. Inclusion criteria were original articles that reported i) the association of genetic variants (SNPs, SNVs and CNVs) of the genes in the DLG protein subfamily or the DLGAP, Shank or Homer protein families with schizophrenia or autism-spectrum disorders; and ii) genetic variations in these genes and their functional consequences, based on animal-model or in vitro studies.
This search initially retrieved 366 articles. After evaluating whether they fulfilled the inclusion criteria, we excluded 261 articles. Another 25 studies and reviews that were relevant for the topic were found from cross-referencing and included in this review. The final pool of articles comprised 130 papers (Fig. 2).

Flow diagram of the literature search. PSD = postsynaptic density.
Results
Table 1 describes SNPs in scaffolding genes that have been associated with schizophrenia or autism-spectrum disorders. Table 2 describes SNVs, and Table 3 describes CNVs. Table 4 describes expression and functional information on scaffolding genes obtained from basic studies. Table 5 describes the SNPs, SNVs and CNVs of risk that are shared by both schizophrenia and autism-spectrum disorders.
Single nucleotide polymorphisms in scaffolding genes associated with schizophrenia, autism-spectrum disorders and other clinical phenotypes of interest
Single nucleotide variants in scaffolding genes associated with schizophrenia, autism-spectrum disorders and other clinical phenotypes of interest
Copy number variants in scaffolding genes associated with schizophrenia, autism-spectrum disorders and other clinical phenotypes of interest
Expression, animal model and pharmacological studies on the reviewed scaffolding genes
Summary of variants in scaffolding genes associated with both schizophrenia and autism-spectrum disorders
DLG protein subfamily
Protein DLG1
Both SNPs in DLG1 have been associated with schizophrenia (Table 1). An SNV has been associated with autism-spectrum disorders (Table 2). Furthermore, deletions of the chromosomal region 3q29 have been related to schizophrenia and autism-spectrum disorders (Table 3) and have been associated with impaired cognition and social dysfunction (Table 3).
Two studies reported reduced expression of DLG1 in the prefrontal cortex of postmortem brain samples of patients with schizophrenia (Table 4). To the best of our knowledge, there are no expression studies in autism-spectrum disorders samples.
An animal-model study reported that glutamate-receptor NMDA antagonists upregulate DLG1 mRNA expression in the cerebral cortex of mice (Table 4).
Protein DLG2
Different studies have identified rare mutations in the DLG2 gene in schizophrenia and autism-spectrum disorders. While SNVs have been detected only in schizophrenia (Table 2), CNVs have been identified in both disorders (Table 3). Interestingly, a deletion identified in intron 6 of the gene in patients with autism-spectrum disorders157 partially overlaps with another deletion spanning from intron 2 to intron 6 in patients with schizophrenia.57
Although still not tested in patients with autism-spectrum disorders, alterations in mRNA and protein expression have been reported in the prefrontal cortex of postmortem brain samples of patients with schizophrenia (Table 4).
Animal-model studies seem to support the function of DLG2 as a regulator of synaptic plasticity; they have shown that DLG2 mutant mice display cognitive abnormalities and long-term potentiation deficits (Table 4).
Protein DLG3
To our knowledge, only 1 genetic study has associated 6 intronic SNPs in DLG3 with autism-spectrum disorders (Table 1), although their consequences for protein function or expression are unknown.
Despite conflicting results in postmortem brain expression studies, it seems that alterations in DLG3 could underlie the neurobiology of schizophrenia. In this regard, both increased and decreased DLG3 mRNA and protein expression have been reported in the thalamus of schizophrenia patients (Table 4).
Animal-model studies support a role for this gene in cognition; mice lacking DLG3 exhibit impaired learning (Table 4).
Protein DLG4
For the DLG4 gene, SNPs (Table 1) and CNVs (Table 3) have been identified only in patients with schizophrenia and with autism-spectrum disorders, respectively. In contrast, SNVs have been associated with both disorders (Table 2).
Among these variants, the SNP rs13331 (T/C), located at the 3′UTR of the gene, is especially interesting because the T allele was first associated with schizophrenia, and a posterior reporter gene assay indicated that subjects carrying this allele had decreased DLG4 protein activity. Based on these results, the authors suggested that reduced DLG4 activity or expression may increase the risk of developing schizophrenia.112
Alterations in mRNA and protein expression have been reported in postmortem brain samples of patients with schizophrenia (Table 4), although the direction of the results is inconsistent. Up to now, no expression studies have been performed in patients with autism-spectrum disorders.
Animal-model studies also appear to support an important role for DLG4 in regulating excitatory synapses and synaptic plasticity, while mutant mice also displayed clinical phenotypes related to schizophrenia and autism-spectrum disorders, such as impaired learning, abnormal communication, altered motor coordination or other abnormal behaviour (Table 4).
Summary
These findings suggest that mutations in DLG genes might increase the risk of developing schizophrenia and autism-spectrum disorders, as well as related cognitive deficits, by contributing to the disruption of glutamatergic synapses. Although the neurobiological mechanisms underlying these disruptions are still unknown, some pathways can be inferred. For instance, mutations affecting DLG2 might modify the tyrosine phosphorylation-based regulation of NMDA receptors, altering NMDA-receptor-related signalling. Similarly, mutations in DLG4 might dysregulate NMDA receptor activity, because this protein also anchors different protein tyrosine kinases.229 Other mechanisms might explain the association between DLG4 and both schizophrenia and autism-spectrum disorders. It has been reported that DLG4 inhibits the interaction between dopamine receptor D1 and the NMDA receptor, preventing a reciprocal damaging overactivation of both receptors.86 This suggests that reduced expression or dysfunction of this protein might dysregulate glutamatergic and dopaminergic homeostasis. Finally, since DLG4 enhances the expression of NMDA receptor subunits NR2A and NR2B230 and the traffic of the NMDA receptor to synapses,231 diminished expression or alterations in protein function might also compromise NMDA-receptor-mediated signalling transduction.
The DLGAP protein family
Some SNPs (Table 1) and SNVs (Table 2) in the DLGAP1 gene and SNVs (Table 2) in the DLGAP3 gene have been associated with schizophrenia. No studies have assessed the association between DLGAP4 and schizophrenia or autism-spectrum disorders.
There is more evidence for an association between the DLGAP2 gene and both disorders. The SNPs (Table 1), SNVs (Table 2) and CNVs (Table 3) in this gene have been associated with both schizophrenia and autism-spectrum disorders. Interestingly, some variants were coincident in both disorders (Table 1, Table 2 and Table 3).
On one hand, the SNP rs2906569 (A>G) in intron 1 and the missense SNP rs2301963 (C>A; P384Q) in exon 3 have been associated with both autism-spectrum disorders115 and schizophrenia.116 Although the functional significance of rs2906569 is difficult to infer, it could affect either the final protein function or the regulation of gene expression by altering different processes, such as splicing, translation regulation or mRNA polyadenylation.232 The missense variant rs2301963, in which CC homozygotes were overrepresented in patients with schizophrenia and patients with autism-spectrum disorders, could affect final protein activity according to bioinformatics analyses.115
On the other hand, 3 nonsynonymous exonic de novo variants (c.841C>G, c.2135C>T and c.2750C>T) have been identified in both schizophrenia116 and autism spectrum disorders.115 The c.841C>G and c.2750C>T mutations were predicted to damage protein function using PolyPhen-2 or Pmut.
Moreover, deletion of the chromosomic region 8p23.3 has been detected in patients with schizophrenia and patients with autism-spectrum disorders. This deletion and other CNVs spanning this gene have been found in patients with schizophrenia and patients with autism-spectrum disorders who have intellectual disability.40,161
To the best of our knowledge, there are no studies of the expression of DLGAP2 in either autism-spectrum disorders or schizophrenia. Animal-model studies have suggested that alterations in this gene might lead to disadaptative social behaviour (Table 4).
Taken together, these findings suggest that disruptions of this gene might alter the function or expression of DLGAP2 and ultimately dysregulate its interplay with other PSD proteins, which could underlie the development of both schizophrenia and autism-spectrum disorders, as well as the manifestation of related clinical phenotypes, such as abnormal social behaviour. Interestingly, animal studies have found that DLGAP2 is vital for normal synaptic structure and function of the orbitofrontal cortex, a brain region that is implicated in the self-regulation of social-emotional behaviour.233 There is also evidence that the orbitofrontal cortex is disrupted in patients with autism, and animal studies have indicated that a lesion of the orbitofrontal cortex may cause aggressive behaviour.234
The Shank protein family
Protein Shank1
Up to now, most of the SNVs in SHANK1 have been associated with autism-spectrum disorders and not schizophrenia (Table 2). Deletions in SHANK1 have also been associated with autism-spectrum disorders (Table 3), with some detected in patients with pronounced social dysfunction.
No expression studies have been carried out for either schizophrenia or autism-spectrum disorders, but animal-model studies seem to indicate that alterations in SHANK1 could lead to impaired social skills (Table 4).
Protein Shank2
While SNVs in this gene have been identified in patients with schizophrenia and patients with autism-spectrum disorders (Table 2), CNVs have been found only in people with autismspectrum disorders, particularly with respect to intellectual disability or language delay (Table 3).
Among these mutations, the SNV (A1731S) detected in patients with schizophrenia is noteworthy. The authors of a study involving a functional assay in HEK293 cells reported that this variant has a significant effect on the F/G-actin ratio and concluded that diminished actin polymerization could lead to impairments in synapse formation and maintenance by reducing the presynaptic contacts.131
To the best of our knowledge, no expression studies have been performed for this gene. However, animal-model studies suggest that disruption of this gene could lead to the cognitive and social dysfunction associated with schizophrenia or autism-spectrum disorders by altering NMDA receptor function (Table 4).
Protein Shank3
There is accumulating evidence that SHANK3 mutations contribute to the pathology of neurodevelopment disorders. Two SNVs in the SHANK3 gene have been identified in patients with schizophrenia (Table 2), and different variants (SNPs, SNVs and CNVs) have been associated with autismspectrum disorders (Table 1, Table 2 and Table 3). Among them, the missense variant G1011V (g.49506159G>T) located in exon 21 has been identified in patients with schizophrenia and patients with autism-spectrum disorders (Table 2). Further studies are needed to test whether this variant has any consequence for the protein function.
The R1117X nonsense mutation (g.49484091C>T) has been identified in exon 21 of the SHANK3 gene in patients with schizophrenia and intellectual disability. This amino acid change resulted in a truncated form of the Shank3 protein that lacked the Homer-binding site, causing its loss of function.46,135
The A198G (g.51117341C>G) identified in people with autism-spectrum disorders generated a frameshift mutation that introduced a premature STOP codon at position 1227, leading to a truncated form of Shank3 that also caused its loss of function. This mutation disrupted actin polymerization, the regulation of spine formation and the molecular organization of the PSD.136
Two frameshift mutations causing premature STOP codons (g.51117094C>G and g.51160615G>T) resulted in the loss of protein function by losing the C-terminal region of the protein, which is crucial for interactions with other PSD proteins.137
Regarding CNVs, different studies have identified deletions in the SHANK3 gene in patients with schizophrenia and patients with autism-spectrum disorders with intellectual disability, developmental delay, language alterations or impaired social interaction (Table 3). Similarly, Phelan McDermid Syndrome (22q13.3 deletion syndrome), which includes deletion of the SHANK3 gene, is characterized by neonatal hypotonia, global developmental delay, absence of speech, autistic behaviour and intellectual disability.235
Animal-model studies also appear to support a role for this gene in the cognitive and behavioural clinical phenotypes related to schizophrenia and autism-spectrum disorders. Shank3 mutant mice display reduced social interaction, affiliation behaviour, repetitive behaviour and communicative deficits (Table 4).
Summary
In summary, results for Shank proteins suggest that their disruption might underlie some of the cognitive and social dysfunction present in schizophrenia and autism-spectrum disorders. This is in line with the latest data showing that the prevalence of SHANK3 mutations in people with autismspectrum disorders is 0.5% to 0.7%, and that a SHANK3 mutation is present in approximately 2% of people with both an autism-spectrum disorder and intellectual disability.129,138,236 More specifically, from animal-model studies it has been suggested that these dysfunctions might be caused by alterations in the NMDA-receptor-related signalling pathway. There is evidence that SHANK2 (−/−) mutant mice display abnormal NMDA receptor function and show alterations in behaviour and social skills.201 Interestingly, when mutant mice were stimulated with NMDA receptor agonists, NMDA receptor function was normalized and their social interactions improved.202 Regarding the pathophysiological mechanisms underlying the social deficits in SHANK3 mutation, it has been reported that knockdown of the SHANK3 gene in rat cortical cultures causes a loss of NMDA receptor function and alterations in its membrane trafficking through the disruption of the actin cytoskeleton.237 Furthermore, Arons and colleagues showed that the loss of function of the SHANK3 gene resulted in reduced glutamatergic synaptic transmission, whereas overexpression of this gene increased the number and size of excitatory synapses and the expression levels of other PSD proteins, such as DLG4 and Homer1.204 Therefore, the behavioural and cognitive alterations present in patients carrying mutations in SHANK genes might be related to dysfunctions in NMDA-receptor-related glutamatergic signalling, which in turn might be caused by abnormalities in the interactions between Shank and other PSD proteins or anomalies in the actin polymerization processes.
The Homer protein family
Although 1 study reported a putative role for the HOMER2 gene in schizophrenia (Table 1), it is principally the HOMER1 gene that has been associated with schizophrenia and autism-spectrum disorders.
Three SNPs in HOMER1 have been associated with schizophrenia (Table 1). Among them, rs4704560 has also been associated with the risk of developing psychotic symptoms in Parkinson disease (Table 1). In addition, 1 SNV and 1 CNV have been found in patients with schizophrenia (Table 2 and Table 3). As well, SNVs (Table 2) and CNVs (Table 3) have been detected in people with autism-spectrum disorders.
Expression studies have reported increases in Homer1a protein and decreases in Homer1b in the hippocampus of postmortem brain samples of schizophrenia (Table 4).
Animal-model studies suggest that HOMER1 transcripts might control cognitive and behaviour functions (Table 4). It has also been suggested that mutations in HOMER1 might increase the risk of developing schizophrenia by dysregulating NMDA receptors and their associated signalling pathway. Pharmacological studies have shown that the NMDR inhibitor phencyclidine (PCP) and the NMDA receptor antagonist ketamine increase HOMER1 mRNA in rat prefrontal cortex and ventral striatum and nucleus accumbens, respectively. Other studies show that HOMER1 mRNA and/or related protein expression levels are modified by psychotomimetic drugs and the antipsychotic haloperidol (Table 3).
Summary
Overall, although HOMER1 has been associated with both schizophrenia and autism-spectrum disorders, genetic, expression and animal model studies do not provide conclusive results. This could be because different Homer1 isoforms have different functions. It has been suggested that the long isoform Homer1c is implicated in the regulation of working memory and sensorimotor function, whereas Homer1a could modulate the behavioural and emotional area.217 Additionally, some studies report that the balance between long and short Homer forms determines the normal functioning of the synaptic architecture and function and influences synaptic plasticity dynamics238; therefore, an alteration of this balance could dysregulate synaptic signalling, leading to neurochemical, structural and behavioural changes.217,218
Discussion
Accumulating evidence supporting biological overlap between schizophrenia and autism-spectrum disorders has fuelled research into common underlying mechanisms to provide a better understanding of the etiology of these disorders, their diagnosis and treatment. One such mechanism involves the synaptic plasticity in which the PSD structure plays a key role. This review has summarized genetic variants in the main scaffolding genes of the PSD that have been associated with schizophrenia and/or autism-spectrum disorders to date. Moreover, evidence coming from genetic, brain expression and animal model studies suggests that genetic variants in scaffolding genes could contribute to the deregulation of the glutamate receptor signalling pathways of the PSD, which may be involved in the pathophysiology of schizophrenia and autism-spectrum disorders, and the development of related shared phenotypes, such as cognitive or social dysfunction.
Such a cross-disorder effect of scaffolding gene and protein dysregulation seems consistent with their role as a dynamic complex that regulates cell signalling pathways and determines the specificity of information flow in intracellular networks.72 Because scaffolding proteins coordinate the excitatory synaptic transmission and mediate functional changes at the synapse, thus regulating synaptic plasticity among other processes, 73 they can be seen as crucial pieces of the complex puzzle of synaptic homeostasis maintenance. The fact that common (SNPs) and rare (SNVs and CNVs) variants have been identified in scaffolding genes in both schizophrenia and autism-spectrum disorders is in agreement with the view that both kinds of variants complementarily and heterogeneously underlie the shared genetic susceptibility to these disorders (Table 5) by generating synaptic instability.
From the present review, it is possible to infer that patients with schizophrenia or autism-spectrum disorders primarily share CNVs that include the complete length of one or more genes. In this regard, CNVs in the PSD genes seem to increase the risk of developing either schizophrenia or autism-spectrum disorders. For instance, deletions of the chromosomal region 3q29, which includes the DLG1 gene, have been related to both schizophrenia and autism-spectrum disorders.45,49 Taking into account the usually large effect size of rare variants on a phenotype, we should not be surprised that alterations in the number of copies (deletions or duplications) of these genes have an impact on PSD functioning and plasticity. In addition to being associated with specific disorders, these variants have been associated with certain phenotypes in these or other disorders, or even in healthy controls. Pocklington and colleagues showed that rare CNV burden may be relevant to cognitive dysfunction in patients with schizophrenia,239 and Stefansson and colleagues found that CNVs conferring risk of either schizophrenia or autism-spectrum disorders, including CNVs in the DLG1 and DLG2 genes, also affect cognitive function in healthy controls.240 Other studies have similarly detected that mutations in PSD genes, including some of the scaffolding genes reviewed here, such as DLG383 or SHANK3,241 are present in patients with intellectual disability.
This review has also provided evidence that, although several SNPs and SNVs in the scaffolding genes have been associated with schizophrenia or autism-spectrum disorders, only a few have been reported in both: 2 SNPs and 3 SNVs in the DLGAP2 gene and 1 SNV in the SHANK3 gene (Table 5). Variants that occur in both diagnoses might be targets of special interest for our understanding of common pathophysiological mechanisms and shared clinical features. Although it is difficult to infer the functional significance of these variants, bioinformatic analyses have indicated that some of the DLGAP2 gene variants (rs2301963, c.841C>G and c.2750C>T) might affect final protein function or expression. In relation to SHANK3, to our knowledge, there is no available information about the functionality of the missense SNV (G1011V) that has also been found associated with both disorders.
Nevertheless, the general lack of specificity observed here can be explained in terms of the pleiotropic nature of scaffolding genes. Variants in different scaffolding genes, either at the allelic or the gene level, may dysregulate the homeostasis of the PSD, which is finally expressed as features associated with different neurodevelopment disorders. In addition to this pleiotropy, the polygenic nature of psychiatric disorders and the polygenic nature of the intermediate molecular pathways known to underlie at least part of the autism-spectrum disorders/schizophrenia pathology (such as the proper functioning of the PSD) should also be considered. This directly links with additional genetic phenomena, such as gene–gene interactions. In recent years, the gene-pathways methodology has been developed to study whether different genes with similar functions are jointly associated with a single phenotype. So far, only a few studies have assessed the effect of common variance in scaffolding genes as a functional gene set or the epistatic effects of other related PSD functional gene sets on the risk of schizophrenia or autism-spectrum disorders. One recent study explored the enrichment of schizophrenia-associated ultra-rare variants and found a significant enrichment of disrupting ultrarare variants among genes defined as encoding interactors with DLG4 and ARC and NMDA receptors.70 Another study observed an enrichment of SNPs associated with autismspectrum disorders in gene sets related to synaptic structure and function, including genes related to scaffolding proteins, β-catenin nuclear pathways, glutamate receptor activity and adherents junctions.67 In addition, although not significant after correction, a nominal association between a PSD protein defined gene set (including ARC and NMDA receptor complexes) with schizophrenia has been reported.36 In all, the effect of common and rare variants in scaffolding genes on schizophrenia and autism-spectrum disorders reflects the complex and heterogeneous genetic architecture of these disorders, and further analyses of gene sets could facilitate the untangling of this complexity.
In addition to genetic data, expression and animal-model studies have indicated the importance of scaffolding genes in schizophrenia and autism-spectrum disorders. There is evidence that patients with schizophrenia or autism-spectrum disorders display deviations from normal scaffolding protein brain expression levels, supporting the hypothesis that the deregulation of these genes might underlie the neurobiology of both disorders. However, to our knowledge, there are no brain expression studies of the 2 genes (DLGAP2 and SHANK3) in which overlapping variants that predispose individuals to schizophrenia and/or autismspectrum disorders were found.52,115,116,135,242 Further research is required to test whether these coincident genetic variants contribute to modifying protein expression levels. In contrast, studies with animal models have shown the importance of scaffolding genes in ensuring cognitive and social function. We have reviewed different studies in which mice with scaffolding protein mutations show schizophrenia and autistim-like phenotypes.184,205 The use of animal models is extremely useful for understanding how changes in the gene sequence can affect phenotypes. As an example of the potential importance of animal models, there are SHANK3-deficient mice in which synaptic deficits were reversed with insulin-like growth factor-1.206 In a recent pilot study, insulin-like growth factor-1 has been used to treat 9 children with autism, and preliminary results have shown a reduction in social deficits.243
Limitations
Some limitations of this review should be acknowledged. First, since the reviewed association studies do not always include the same genetic regions or variants, coincident variants can be linked to shared genetic variability between schizophrenia and autism-spectrum disorders, but noncoincidence cannot be interpreted as a lack of it. Second, patients with low IQ scores are generally excluded from association studies. Since the scaffolding genes reviewed here seem to contribute to cognitive phenotypes, it is plausible to hypothesize that the effects of SNPs and SNVs in these genes were detected less often than they actually occur. Third, the relationship between the effect size associated with common and rare variants, the statistical power needed to detect these effects and the sample sizes in the studies reported in the different articles reviewed should be considered. The odds ratios associated with schizophrenia risk SNPs are typically about 1.10 to 1.50, whereas schizophrenia-associated CNVs confer a significantly increased risk of illness (odds ratios for several CNVs exceed 8).244 Therefore, rare variants can more easily create significant genome-wide associations than common variants.245
Conclusion
Advances in genetic technologies, together with the assembly of large patient cohorts, have made it possible to identify some genes and biological pathways involved in both schizophrenia and autism-spectrum disorders. Among them, scaffolding genes implicated in the PSD have been repeatedly associated with schizophrenia and autism-spectrum disorders, pointing toward these genes’ common involvement in the neurobiology of these disorders and in some shared clinical phenotypes, such as cognitive and social impairment. This review summarizes evidence that many different variants could introduce numerous slight alterations in the PSD pathway, leading to its inappropriate development or insufficiently robust response to environmental insults.
Acknowledgements
This study was supported by: the Network of European Funding for Neuroscience Research, ERA-NET NEURON (PiM2010ERN-00642); Instituto de Salud Carlos III through the project PI15/01420 (co-funded by European Regional Development Fund /European Social Fund, “Investing in your future”); and SAF 2015-71526-REDT; and Ajuts de Personal Investigador predoctoral en Formació (APIF) to J Soler, 2017-2018. Thanks to the Comissionat per a Universitats i Recerca del DIUE (2014SGR1636).
Footnotes
Competing interests: M. Parellada reports personal fees from Fundación Orange, outside the submitted work. M.-O. Krebs reports personal fees or travel grant from Janssen and Otsuka-Lundbeck, outside the submitted work. No other competing interests declared.
Contributors: J. Soler and M. Fatjó-Vilas designed the study and acquired the data, which all authors analzyed. J. Soler and M. FatjóVilas wrote the article, which all authors reviewed. All authors approved the final version to be published and can certify that no other individuals not listed as authors have made substantial contributions to the paper.
- Received April 5, 2017.
- Revision received October 18, 2017.
- Accepted November 13, 2017.
References

{kind=link}
{kind=link}
Article tools





